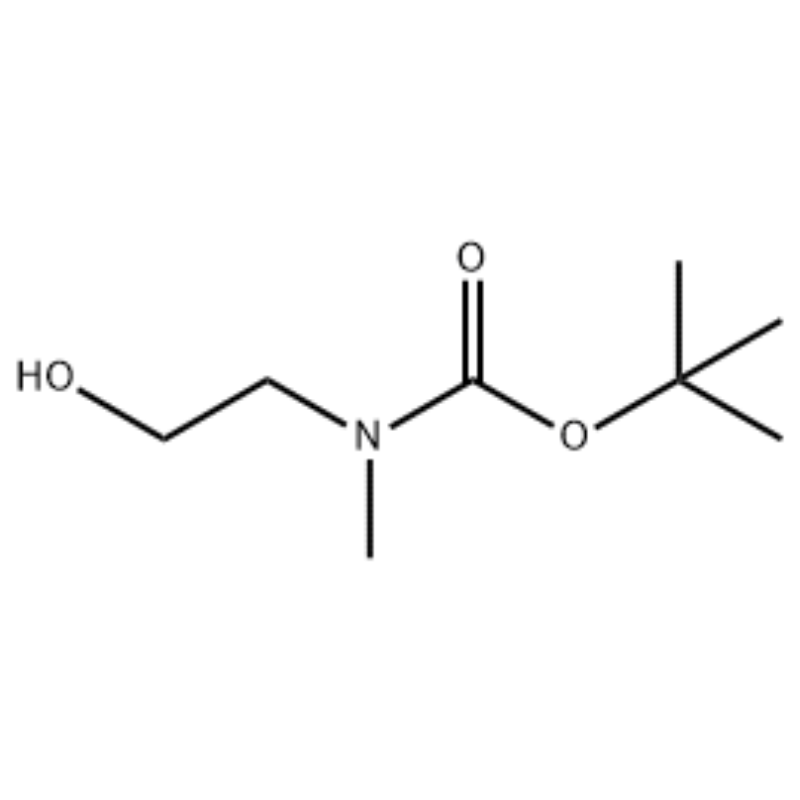
To a solution of 2-(methylamino)ethanol (500 mg, 0.53 ml, 6.66 mmol) in CH2Cl2 (20 ml) was added Boc2O (1.48 g, 6.79 mmol), followed by stirring at room temperature for 1 hour. The reaction solution was extracted with brine and CH2Cl2. The organic layer thus obtained was dried over MgSO4 and filtered. Then, the filtrate was concentrated in vacuo to obtain the object compound (colorless oil, quantitative); 1H NMR (200 MHz, CDCl3) delta 3.74 (q, J= 10.5, 5.2 Hz, 2H) 3.25 (t, J= 5.2 Hz, 2H) 2.91 (s, 3H) 1.45 (s, 9H); mass spectrum m/e (relative intensity) 144 (20) 102 (24) 57 (70) 44 (100).
Example 38; N1-(3-Fluoro-4-(2-(1-(2-(methylamino)ethyl)-1H-imidazol-4-yl)thieno[3,2-b]pyridin-7-yloxy)phenyl)-N3-(2-methoxyphenyl)malonamide (96); Step 1: tert-Butyl 2-hydroxyethyl(methyl)carbamate (97) (J. Med. Chem., 1999, 42, 11, 2008) To a solution of 2-(methylamino)ethanol (5.0 g, 67 mmol) in THF (50 ml) at RT was added Boc2O (15.7 g, 72 mmol) and the reaction mixture was stirred at RT for 4 hours. The reaction mixture was concentrated to dryness and the title compound 97 was used directly in the next step with no additional purification (11.74 g, 100% yield). MS (m/z): 176.2 (M+H).
Preparation of l-2-[4-Bromo-2-(4-oxo-2-ftiotaioxo1hiazolidin-5-ylidenemefliyl)phenoxy]efliyl-3-efliyl-l- methylurea(Compoiotamd 161)Step 1 : Synthesis of t-butyl2-hydroxyethylmethylcarbamate; To a solution of 2-(methylamino)ethanol (500 mg, 0.53 ml, 6.66 mmol) in CH2Cl2 (20 ml) was added BoC2O (1.48 g,6.79 mmol), followed by stirring at room temperature for 1 hour. The reaction solution was extracted with brine and CH2Cl2. The organic layer thus obtained was dried over MgSO4 and filtered. Then, the filtrate was concentrated in vacuo to obtain the object compound (colorless oil, quantitative);1HNMR (200 MHz, CDCl3) delta 3.74 (q, J= 10.5, 5.2 Hz, 2H) 3.25 (t, J= 5.2 Hz, 2H) 2.91 (s, 3H) 1.45 (s, 9H); mass spectrum m/e (relative intensity) 144 (20) 102 (24) 57 (70) 44 (100).
2-(methylamino)ethanol (90.1 g, 1.2 mol) was dissolved in 1.2 L of methylene chloride, and BoC2O (218 g, 1 mol) was slowly added thereto while stirring at 00C, followed by at room temperature for 3 hours. The reaction mixture was sequentially washed with 700 mL of an aqueous solution of saturated ammonium chloride, and 300 mL of water. The washed mixture was dehydrated using anhydrous sodium sulfate and concentrated under a reduced pressure, to obtain the compound (a) (175 g, 1 mol, 100%) as an oil with no color.TLC : Rf = 0.5 (50% EtOAc in Hex) visualized with Ce-Mo stain1H NMR (600MHz, CDCl3) delta 1.47 (s, 9H), 2.88 (br s, IH), 3.41 (br s, 2H), 3.76 (br s, 2H).
90.1 g (1.2 mol) of 2-(methylamino)ethanol was dissolved in 1.2 L of methylene chloride, 218 g (1 mol) of Boc2O was slowly added thereto while the resulting solution was stirred at 0C, and the resulting solution was stirred at room temperature for 3 hours. The reaction mixture was sequentially washed with 700 mL of an aqueous saturated ammonium chloride solution and 300 mL of water, dehydrated using anhydrous sodium sulfate, and then concentrated under reduced pressure to obtain 175 g (1 mol) of an achromic oil compound protected by the Boc group (yield: 100%). [0140] 1H NMR (600MHz, CDCl3) delta 7.84 (br s, 2H), 7.76 (br s, 2H), 4.34 (d, J = 15.0 Hz, 2H), 3.63 (br s, 2H), 3.04 (d, J = 15.0 Hz, 3H), 1.46 (d, J = 16.2 Hz, 9H) [0141] 90 g (0.514 mol) of the obtained compound was dissolved in 1.5 L of tetrahydrofuran, 88.0 g (539 mol) of N-hydroxyphthalimide and 141 g (0.539 mol) of triphenylphosphine were added thereto, 106 mL (0.539 mol) of diisopropyl azodicarboxylate was slowly added thereto while stirring the resulting solution at 0C, and the resulting solution was stirred for 3 hours while the temperature thereof was raised to room temperature. After concentration of the reaction mixture under reduced pressure, 600 mL of isopropylether was added thereto, the resulting solution was stirred at 0C for 1 hour, and white solid-type triphenylphosphine oxide was filtered. The solid was washed with 200 mL of isopropylether cooled to 0C and collected with the first filtrate, and the resulting filtrate was concentrated under reduced pressure to obtain 198 g of a mixture of Compound XX and diisopropyl hydrazodicarboxylate in a mixing ratio of 10 to 15% (yield: 120%). [0142] 1H NMR (600MHz, CDCl3) delta 7.84 (br s, 2H), 7.76 (br s, 2H), 4.34 (d, J = 15.0 Hz, 2H), 3.63 (br s, 2H), 3.04 (d, J = 15.0 Hz, 3H), 1.46 (d, J= 16.2 Hz, 9H)
















 Building 12, No.309, South 2nd Road, Economic Development Zone, Longquanyi District, Chengdu, Sichuan, China.
Building 12, No.309, South 2nd Road, Economic Development Zone, Longquanyi District, Chengdu, Sichuan, China. amy@enlaibio.com / cynthia@enlaibio.com / edison@enlaibio.com / daisy@enlaibio.com
amy@enlaibio.com / cynthia@enlaibio.com / edison@enlaibio.com / daisy@enlaibio.com +86 (028) 84841969
+86 (028) 84841969 +86 135 5885 5404
+86 135 5885 5404



.png)


